Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences
In this work, a Graph Neural Network (GNN) is used to encode the graph structure of a compound (i.e. molecule) into a fixed-size vector. Moreover, a CNN is used to encode a protein’s chain of amino acids (an amino acid is a group of 3 DNA bases) into the same embedding space as the output of the GNN.
Data
The datasets used with this model contain pairs of (compound, protein) and a boolean to indicate if there is an interaction. Compounds are stored as SMILES, a notation that represents their structure as a string. Proteins are chains of amino acids, and since each amino acid has an associated letter, a simple string is used.
Interestingly, proteins are also molecules (compounds), and could technically be represented as SMILES and treated as a graph. However, since they are huge-ass molecules (also called macromolecules), it makes more sense to use an amino acid sequence, which allows to treat them with a CNN.
Model
The full model concatenates the vector representation of a compound and the vector of a protein, and performs a binary classification of whether they will interact or not. (e.g. Will the compound bind to the protein?)
A more complex and effective version of the model uses an attention mechanism as the last layer of the CNN, which can be viewed as using a weighted average instead of a plain global average pooling.
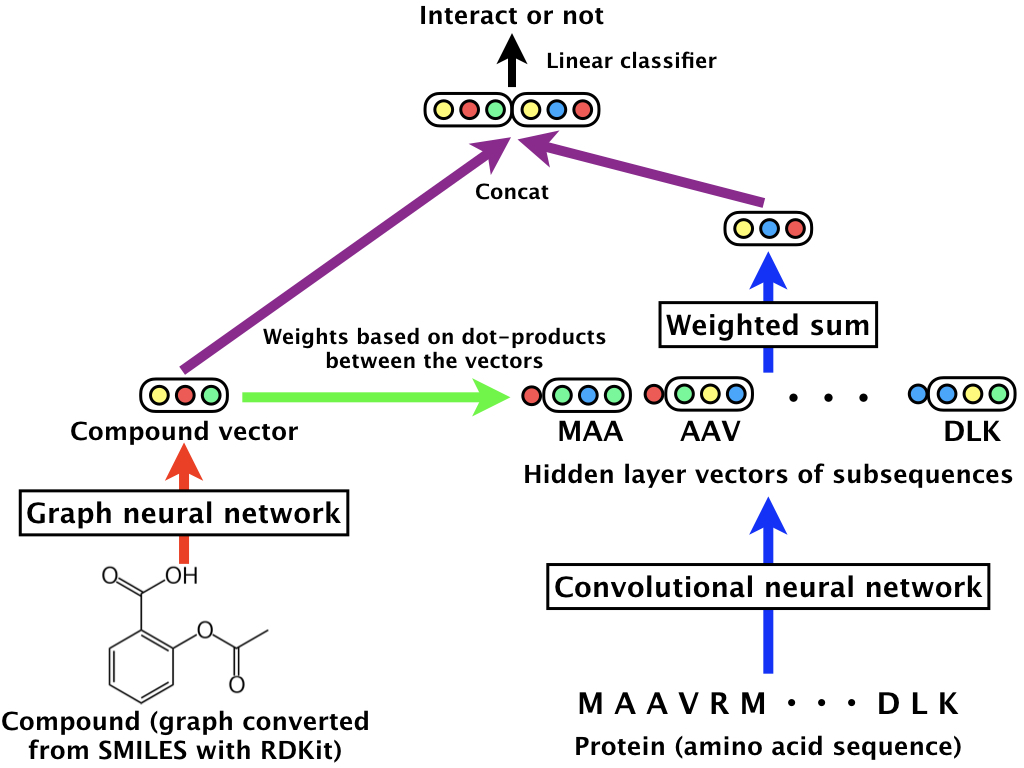
The GNN works in multiple phases:
- Embed the atoms into vertex vectors;
- Apply multiple time steps of vertex transitions and edge transitions;
- Aggregate the vertices by taking the average of their vectors.
Below is a figure representing the vertex transition function:
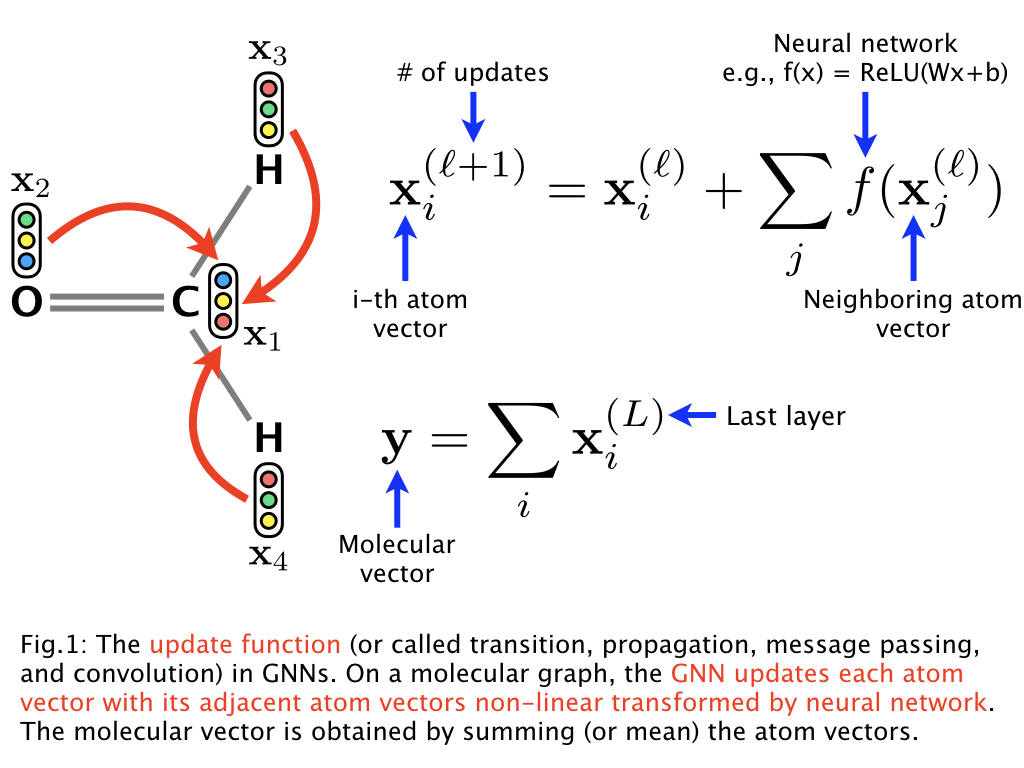
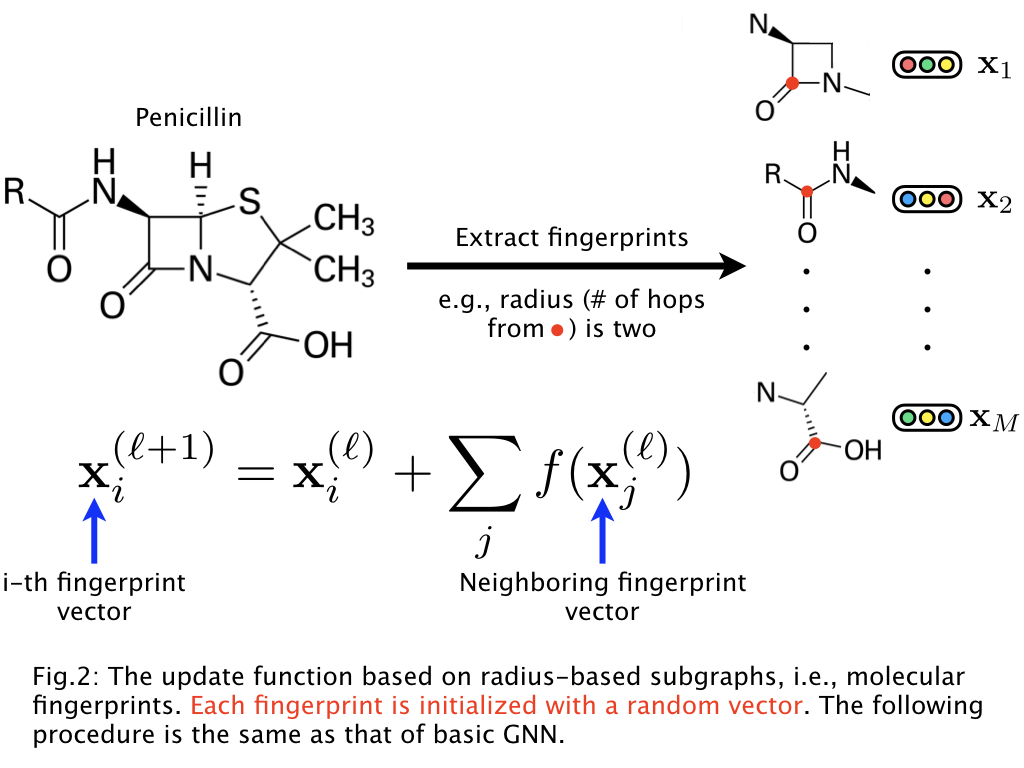
The authors claim that they obtain better results by embedding atoms using a representation of their neighborhood. This method seems to be exactly the same as embedding each atom independently and running multiple vertex transitions. This confusion is not cleared up in the paper.
Results
This approach is compared to other machine learning methods (such as SVM) and non-learned methods. The proposed method is either competitive with others, or outperforms them significantly, depending on the task.
One clear advantage of the method is that its precision and recall are improved by using a higher ratio of negative examples, where in other ML methods these metrics are lowered.
Code available on https://github.com/masashitsubaki/CPI_prediction and https://github.com/masashitsubaki/GNN_molecules. Images taken on github.